
Clinical Evaluation requirements have increased dramatically. The process now involves three documents: the Clinical Development Plan (CDP), Clinical Evaluation Plan (CEP) and Clinical Evaluation Report (CER). The clinical team at Qserve has been busy providing virtual trainings via webinar and writing MEDDEV and MDR compliant CEPs and CERs. This blog will cover a few tips and tricks.
Sufficient Clinical Data Versus Sufficient Clinical Evidence
Many of the CERs that Qserve evaluates have plenty of data. What they often lack is an analysis of the data presented. Data on its own is meaningless without context. Data only becomes evidence when a specific viewpoint is added. The process of clinical evaluation starts with the origin of a research question based on the data at hand. The final step is to evaluate that data to determine if you have sufficient clinical evidence to support conformity to GSPRs 1 and 8 (at a minimum) of the MDR or ERs 1, 3 and 6 of the MDD. Evidence is built by telling the story about the data. For example, if adverse event data from PMS and clinical literature are used together to determine the occurrence rate of clinical risks associated with the device, there is a higher degree of confidence that the information is correct. The two sources analysed together create evidence. If the two sources disagree, more data must be collected. This process is done for each performance and safety requirement, as well as the claims and benefits outlined in the CEP. Data from bench-testing, PMS, clinical investigation, and clinical literature can be analysed together to build sufficient clinical evidence.
How do you get started?
The process of clinical evaluation requires input from many different departments within an organization. The CEP should provide a detailed story as to why the device was designed and what it is designed to do. The MEDDEV Rev 4 provides a great description of how to define the scope of the clinical evaluation based on the Essential Requirements (now General Safety and Performance Requirements under the MDR) that need to be addressed from a clinical perspective and the nature and history of the device in Section 7. The purpose of the plan is to assess the data already available to support the intended use, performance, safety, and benefits of the device.
If you have been assigned to write a clinical evaluation plan and report, your first step is to contact all the relevant departments in your organization to collect the required information for the device. The structure of every manufacturer is different, but your organization will have similar departments to those listed in the table below. The data you will need for a new device is slightly different than what is needed for an already CE marked device in that after CE marking, Post Market Surveillance (PMS) data and Post Market Clinical Follow-up data must be included in the CER.
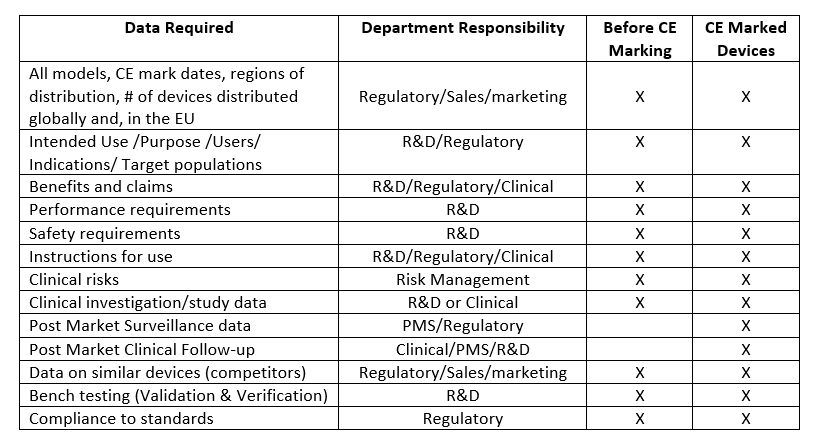
After assessing data available, research questions are created to specifically address gaps in data or to support existing data. Data can then be pulled from clinical literature, device registries, healthcare databases, or disease specific databases to answer the defined research questions. Clinical data from literature is a required first step. The other options listed can be used to enhance the data found or to fill in gaps if data is not available in the literature.
Establishing State-of-the-Art (SoA)
A critical component of the clinical evaluation process is establishing the state-of-the-art in medicine (clinical SoA) for the disease that the device is intended to treat, monitor, or diagnose. The SoA in medicine will change over time, which means that the benefit-risk profile for legacy devices may also change as new devices that address previously accepted risks enter the market. The discussion of SoA should start in the CEP with the background of the disease associated with the device, including all of its forms and severities, as well as, any alternative treatment options, including medicinal treatments. Applicable standards and guidance documents related to the device being evaluated should also be included to provide the technical aspect of SoA.
This discussion provides a framework by which to verify that clinical risks associated with the device being evaluated are comparable to other treatments for the disease in similar patient populations. To that end, the SoA in the CEP should also include acceptance criteria for data on performance, safety, and benefits, as well as, the benefit-risk ratio and acceptability of side-effects evaluated in the clinical evaluation. This will serve as a justification for the amount of clinical evidence necessary to demonstrate conformity with the relevant general safety and performance requirements as required by MDR Article 61, point 1. It will also provide a basis for establishing the device’s place in the global treatment portfolio for the disease as described in MEDDEV 2.7/1 Rev 4 Appendix 7.2.
The SoA in the CER will build on this discussion to include prognosis if the disease if left untreated, and the risks and benefits of the alternate devices discussed in the CEP. Other information required for SoA in the CER includes:
- Frequency of device use in the general population
- Diverging opinions of professionals concerning device use and treatment options
- Unmet needs of device technology
- Clinical hazards (risks) to at risk patients in the general population – pediatric, elderly and pregnant patients.
There are many more aspects of the clinical evaluation process that cannot be captured in a blog. We offer customized virtual trainings via that reviews gaps in an existing CER and provides instruction on how to fill those gaps to meet regulatory requirements. The training is broken down into 5 modules that are roughly 45 minutes in length. We also provide a Q&A session after the training to answer any remaining questions.
The modules are:
- Module 1: Clinical safety, risks and side-effects (including maximizing PMS data)
- Module 2: Clinical performance, determining measurable endpoints and support with clinical data
- Module 3: Risk/benefit (including state-of-the-art)
- Module 4: CEP and equivalency
- Module 5: PMCF requirements under the MDR
We are also available to peer review CERs prior to submission. Please contact Qserve for a quote if you would like customized training and/or peer review to help with clinical data requirements for the EU.